Español
Español
Genomic landscape of transcriptional and epigenetic dysregulation in early-onset polyglutamine disease
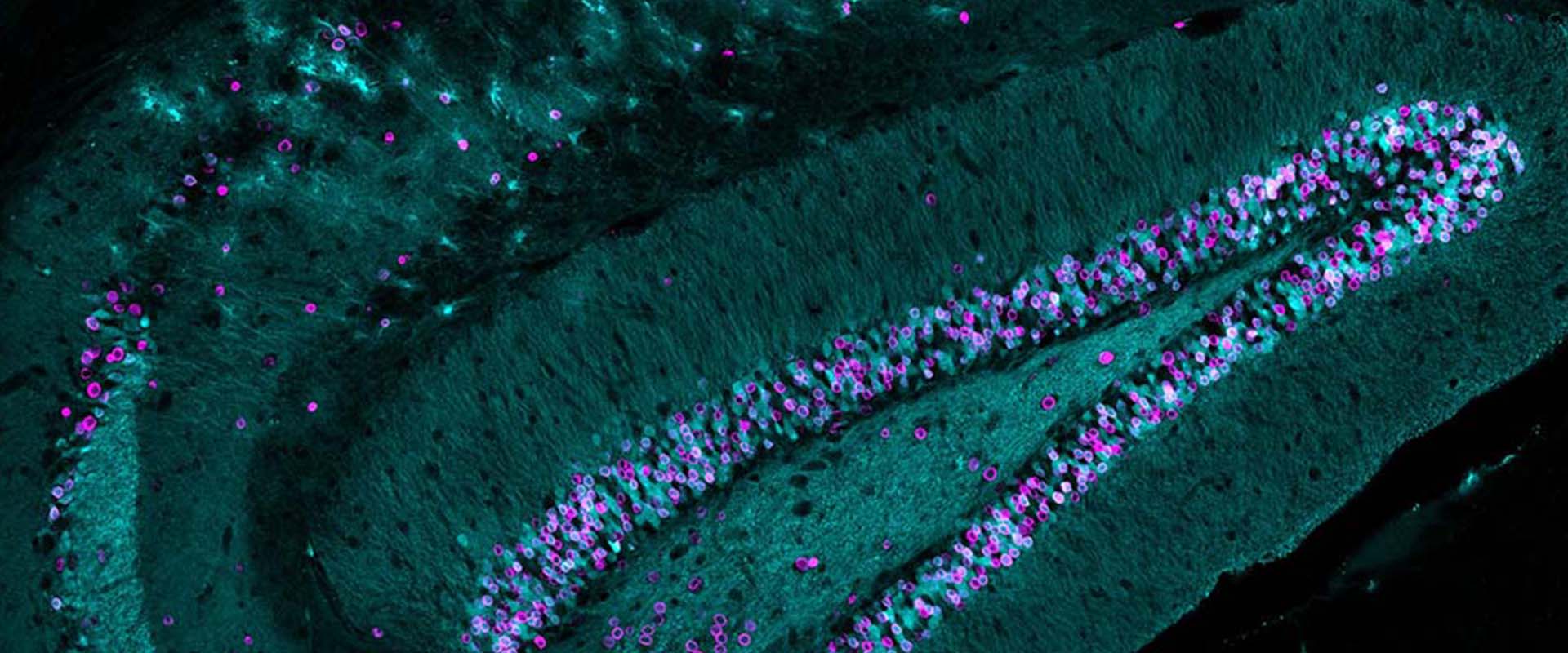
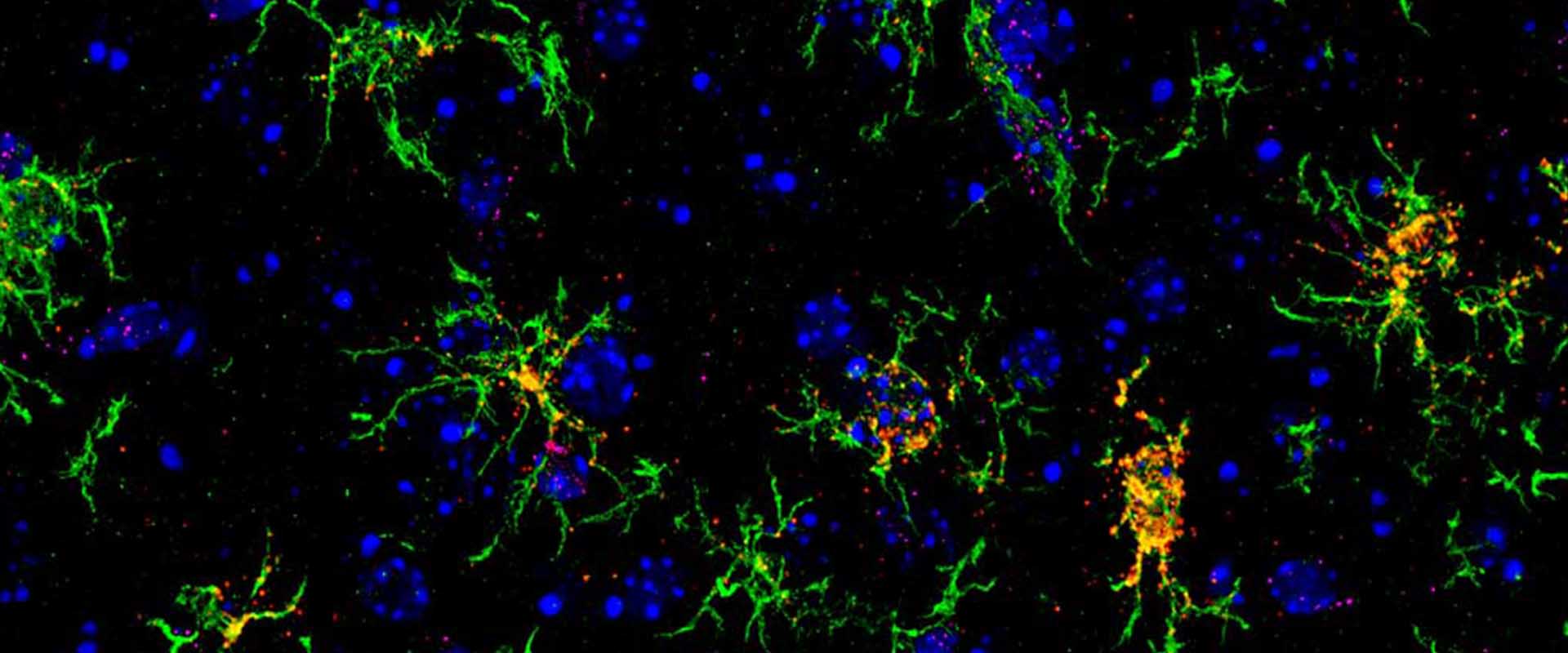
Transcriptional dysregulation is an important early feature of polyglutamine diseases. One of its proposed causes is defective neuronal histone acetylation, but important aspects of this hypothesis, such as the precise genomic topography of acetylation deficits and the relationship between transcriptional and acetylation alterations at the whole-genome level, remain unknown. The new techniques for the mapping of histone posttranslational modifications at genomic scale enable such global analyses and are challenging some assumptions about the role of specific histone modifications in gene expression. We examined here the genome-wide correlation of histone acetylation and gene expression defects in a mouse model of early-onset Huntington’s disease. Our analyses identified hundreds of loci that were hypoacetylated for H3K9,14 and H4K12 in the chromatin of these mice. Surprisingly, few genes with altered transcript levels in mutant mice showed significant changes in these acetylation marks and vice versa. Our screen, however, identified a subset of genes in which H3K9,14 deacetylation and transcriptional dysregulation concur. Genes in this group were consistently affected in different brain areas, mouse models and tissue from patients, which suggests a role in the etiology of this pathology. Overall, the combination of histone acetylation and gene expression screenings demonstrates that histone deacetylation and transcriptional dysregulation are two early, largely independent, manifestations of polyglutamine disease and suggests that additional epigenetic marks or mechanisms are required for explaining the full range of transcriptional alterations associated with this disorder.